Les technologies de transcriptomique sont les techniques utilisées pour étudier le transcriptome d'un organisme , la somme de tous ses transcrits d'ARN . Le contenu informationnel d'un organisme est enregistré dans l'ADN de son génome et exprimé par transcription . Ici, l'ARNm sert de molécule intermédiaire transitoire dans le réseau d'information, tandis que les ARN non codants remplissent diverses fonctions supplémentaires. Un transcriptome capture un instantané dans le temps de l'ensemble des transcrits présents dans une cellule . Les technologies de transcriptomique fournissent un aperçu général des processus cellulaires actifs et dormants. L'un des principaux défis de la biologie moléculaire est de comprendre comment un génome unique donne naissance à une variété de cellules. Un autre défi est de savoir comment l'expression des gènes est régulée.
Les premières tentatives d’étude de transcriptomes entiers ont commencé au début des années 1990. Les avancées technologiques qui ont suivi depuis la fin des années 1990 ont transformé à plusieurs reprises le domaine et ont fait de la transcriptomique une discipline répandue dans les sciences biologiques. Il existe deux techniques contemporaines clés dans le domaine : les microarrays , qui quantifient un ensemble de séquences prédéterminées, et le RNA-Seq , qui utilise le séquençage à haut débit pour enregistrer tous les transcriptomes. Au fur et à mesure que la technologie s’est améliorée, le volume de données produites par chaque expérience de transcriptome a augmenté. En conséquence, les méthodes d’analyse des données ont été progressivement adaptées pour analyser de manière plus précise et plus efficace des volumes de données de plus en plus importants. Les bases de données de transcriptomes deviennent de plus en plus grandes et utiles à mesure que les transcriptomes continuent d’être collectés et partagés par les chercheurs. Il serait presque impossible d’interpréter les informations contenues dans un transcriptome sans la connaissance des expériences précédentes.
La mesure de l'expression des gènes d'un organisme dans différents tissus ou conditions , ou à différents moments, fournit des informations sur la façon dont les gènes sont régulés et révèle des détails sur la biologie d'un organisme. Elle peut également être utilisée pour déduire les fonctions de gènes non annotés auparavant . L'analyse du transcriptome a permis d'étudier la façon dont l'expression des gènes change dans différents organismes et a joué un rôle déterminant dans la compréhension des maladies humaines . Une analyse de l'expression des gènes dans son intégralité permet de détecter de larges tendances coordonnées qui ne peuvent pas être discernées par des analyses plus ciblées .
Histoire
La transcriptomique a été caractérisée par le développement de nouvelles techniques qui ont redéfini ce qui est possible tous les dix ans environ et ont rendu les technologies précédentes obsolètes. La première tentative de capture d'un transcriptome humain partiel a été publiée en 1991 et a rapporté 609 séquences d'ARNm du cerveau humain . En 2008, deux transcriptomes humains, composés de millions de séquences dérivées de transcriptions couvrant 16 000 gènes, ont été publiés, et en 2015, des transcriptomes avaient été publiés pour des centaines d'individus. Des transcriptomes de différents états pathologiques , tissus ou même cellules individuelles sont désormais générés de manière routinière. Cette explosion de la transcriptomique a été alimentée par le développement rapide de nouvelles technologies avec une sensibilité et une économie améliorées.
Avant la transcriptomique
Des études sur des transcriptions individuelles ont été réalisées plusieurs décennies avant que des approches transcriptomiques ne soient disponibles. Des bibliothèques de transcriptions d'ARNm de vers à soie ont été collectées et converties en ADN complémentaire (ADNc) pour être stockées à l'aide de la transcriptase inverse à la fin des années 1970. Dans les années 1980, le séquençage à faible débit utilisant la méthode Sanger a été utilisé pour séquencer des transcriptions aléatoires, produisant des étiquettes de séquence exprimée (EST). La méthode de séquençage de Sanger était prédominante jusqu'à l'avènement des méthodes à haut débit telles que le séquençage par synthèse (Solexa/Illumina). Les EST sont devenues importantes dans les années 1990 en tant que méthode efficace pour déterminer le contenu génétique d'un organisme sans séquencer l'ensemble du génome . Les quantités de transcriptions individuelles ont été quantifiées à l'aide de méthodes de transfert Northern , de matrices de membranes en nylon et plus tard de PCR quantitative à transcriptase inverse (RT-qPCR), mais ces méthodes sont laborieuses et ne peuvent capturer qu'une minuscule sous-section d'un transcriptome. Par conséquent, la manière dont un transcriptome dans son ensemble est exprimé et régulé est restée inconnue jusqu'à ce que des techniques à plus haut débit soient développées.
Premières tentatives
Le terme « transcriptome » a été utilisé pour la première fois dans les années 1990. En 1995, l'une des premières méthodes transcriptomiques basées sur le séquençage a été développée, l'analyse en série de l'expression génétique (SAGE), qui fonctionnait par séquençage Sanger de fragments de transcription aléatoires concaténés. Les transcriptions ont été quantifiées en faisant correspondre les fragments à des gènes connus. Une variante de SAGE utilisant des techniques de séquençage à haut débit, appelée analyse numérique de l'expression génétique, a également été brièvement utilisée. Cependant, ces méthodes ont été largement dépassées par le séquençage à haut débit de transcriptions entières, qui a fourni des informations supplémentaires sur la structure de la transcription telles que les variantes d'épissage .
Développement de techniques contemporaines
Français Les techniques contemporaines dominantes, les microarrays et le RNA-Seq , ont été développées au milieu des années 1990 et 2000. Les microarrays qui mesurent l'abondance d'un ensemble défini de transcrits via leur hybridation à un réseau de sondes complémentaires ont été publiés pour la première fois en 1995. La technologie des microarrays a permis de tester des milliers de transcrits simultanément et à un coût par gène considérablement réduit et en économisant du travail. Les réseaux d'oligonucléotides tachetés et les réseaux haute densité Affymetrix étaient la méthode de choix pour le profilage transcriptionnel jusqu'à la fin des années 2000. Au cours de cette période, une gamme de microarrays a été produite pour couvrir les gènes connus dans des organismes modèles ou économiquement importants. Les progrès dans la conception et la fabrication des réseaux ont amélioré la spécificité des sondes et ont permis de tester davantage de gènes sur un seul réseau. Les progrès dans la détection par fluorescence ont augmenté la sensibilité et la précision de mesure pour les transcrits à faible abondance.
Français L'ARN-Seq est accompli par la transcription inverse de l'ARN in vitro et le séquençage des ADNc résultants . L'abondance des transcrits est dérivée du nombre de comptages de chaque transcrit. La technique a donc été fortement influencée par le développement des technologies de séquençage à haut débit . Le séquençage de signatures massivement parallèles (MPSS) était un des premiers exemples basé sur la génération de séquences de 16 à 20 pb via une série complexe d' hybridations , et a été utilisé en 2004 pour valider l'expression de dix mille gènes chez Arabidopsis thaliana . Les premiers travaux sur l'ARN-Seq ont été publiés en 2006 avec cent mille transcrits séquencés à l'aide de la technologie 454. Cette couverture était suffisante pour quantifier l'abondance relative des transcrits . L'ARN-Seq a commencé à gagner en popularité après 2008 lorsque les nouvelles technologies Solexa/Illumina ont permis d'enregistrer un milliard de séquences de transcrits. Ce rendement permet désormais la quantification et la comparaison des transcriptomes humains.
Collecte de données
La génération de données sur les transcrits d'ARN peut être réalisée via l'un des deux principes principaux : le séquençage de transcrits individuels ( EST ou RNA-Seq) ou l'hybridation de transcrits à un réseau ordonné de sondes nucléotidiques (microarrays).
Isolement de l'ARN
Toutes les méthodes transcriptomiques nécessitent que l'ARN soit d'abord isolé de l'organisme expérimental avant que les transcriptions puissent être enregistrées. Bien que les systèmes biologiques soient incroyablement divers, les techniques d'extraction d'ARN sont globalement similaires et impliquent la rupture mécanique des cellules ou des tissus, la rupture de la RNase avec des sels chaotropiques , la rupture des macromolécules et des complexes nucléotidiques, la séparation de l'ARN des biomolécules indésirables , y compris l'ADN, et la concentration de l'ARN par précipitation à partir d'une solution ou élution à partir d'une matrice solide . L'ARN isolé peut en outre être traité avec de la DNase pour digérer toute trace d'ADN. Il est nécessaire d'enrichir l'ARN messager car les extraits d'ARN totaux sont généralement constitués à 98 % d'ARN ribosomique . L'enrichissement des transcriptions peut être effectué par des méthodes d'affinité poly-A ou par déplétion de l'ARN ribosomique à l'aide de sondes spécifiques à la séquence. L'ARN dégradé peut affecter les résultats en aval ; Par exemple, l'enrichissement en ARNm à partir d'échantillons dégradés entraînera l'épuisement des extrémités 5' de l'ARNm et un signal irrégulier sur toute la longueur d'un transcrit. La congélation instantanée des tissus avant l'isolement de l'ARN est typique, et des précautions sont prises pour réduire l'exposition aux enzymes RNase une fois l'isolement terminé.
Balises de séquences exprimées
Une séquence EST ( Exprimed Sequence Tag ) est une courte séquence nucléotidique générée à partir d'un seul transcrit d'ARN. L'ARN est d'abord copié sous forme d'ADN complémentaire (ADNc) par une enzyme transcriptase inverse avant que l'ADNc résultant ne soit séquencé. Étant donné que les EST peuvent être collectées sans connaissance préalable de l'organisme dont elles proviennent, elles peuvent être fabriquées à partir de mélanges d'organismes ou d'échantillons environnementaux. Bien que des méthodes à haut débit soient désormais utilisées, les bibliothèques EST fournissaient généralement des informations de séquence pour les premières conceptions de puces à ADN ; par exemple, une puce à ADN d'orge a été conçue à partir de 350 000 EST précédemment séquencés.
Analyse en série et en cap de l'expression des gènes (SAGE/CAGE)
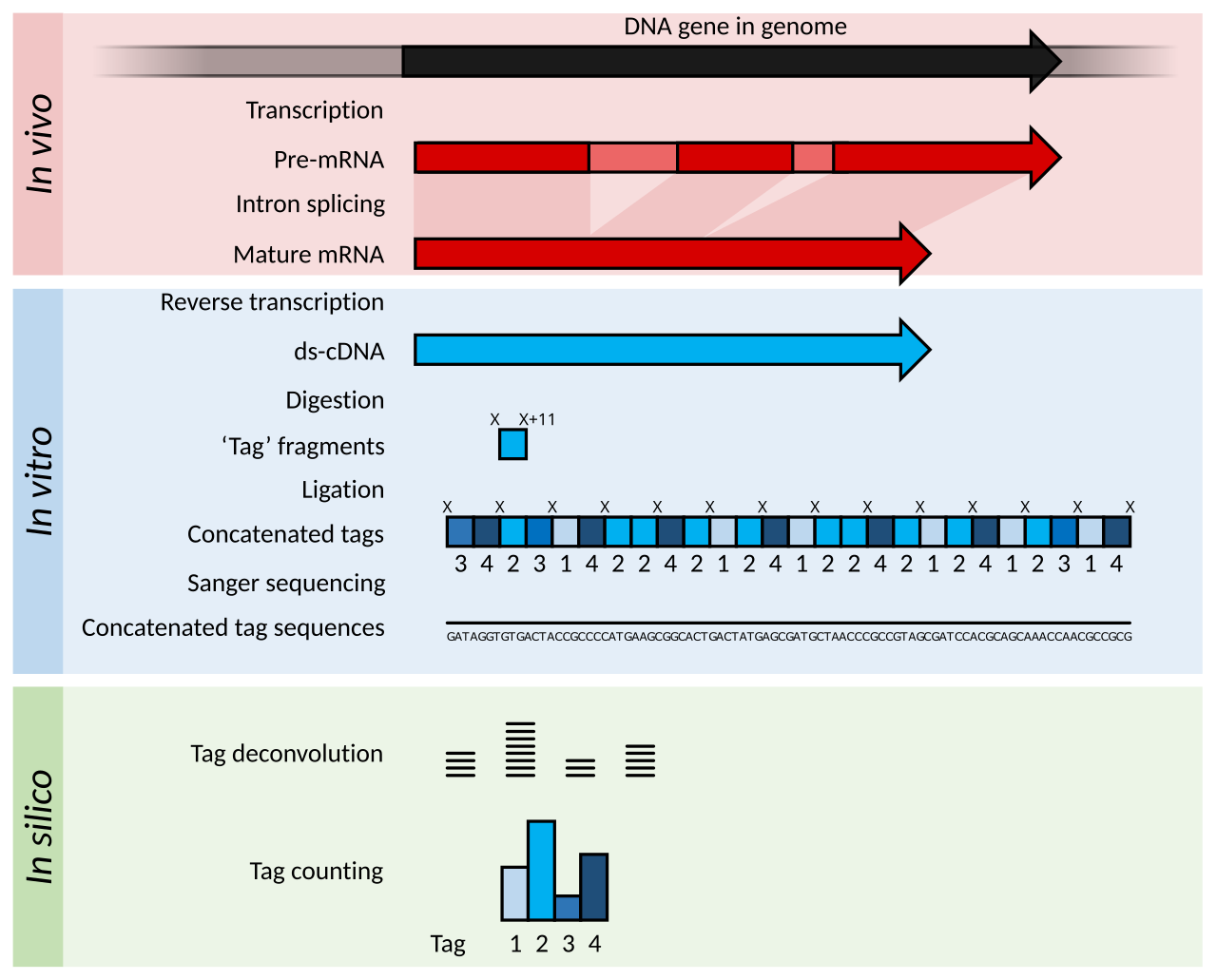
L'analyse en série de l'expression génétique (SAGE) est un développement de la méthodologie EST pour augmenter le débit des étiquettes générées et permettre une certaine quantification de l'abondance des transcriptions. L'ADNc est généré à partir de l' ARN, mais est ensuite digéré en fragments « d'étiquettes » de 11 pb à l'aide d'enzymes de restriction qui coupent l'ADN au niveau d'une séquence spécifique et de 11 paires de bases le long de cette séquence. Ces étiquettes d'ADNc sont ensuite jointes tête-bêche en longs brins (> 500 pb) et séquencées à l'aide de méthodes à faible débit, mais à longue longueur de lecture, telles que le séquençage de Sanger . Les séquences sont ensuite divisées en leurs étiquettes d'origine de 11 pb à l'aide d'un logiciel informatique dans un processus appelé déconvolution . génome de référence de haute qualité est disponible, ces étiquettes peuvent être associées à leur gène correspondant dans le génome. Si un génome de référence n'est pas disponible, les étiquettes peuvent être directement utilisées comme marqueurs diagnostiques si elles s'avèrent être exprimées de manière différentielle dans un état pathologique.
La méthode d' analyse de l'expression génique par cap analysis (CAGE) est une variante de SAGE qui séquence les marqueurs à partir de l' extrémité 5' d'un transcrit d'ARNm uniquement. Par conséquent, le site de démarrage de la transcription des gènes peut être identifié lorsque les marqueurs sont alignés sur un génome de référence. L'identification des sites de démarrage des gènes est utile pour l'analyse des promoteurs et pour le clonage d'ADNc complets.
Les méthodes SAGE et CAGE produisent des informations sur plus de gènes que ce qui était possible lors du séquençage d'EST simples, mais la préparation des échantillons et l'analyse des données nécessitent généralement plus de travail.
Microarrays
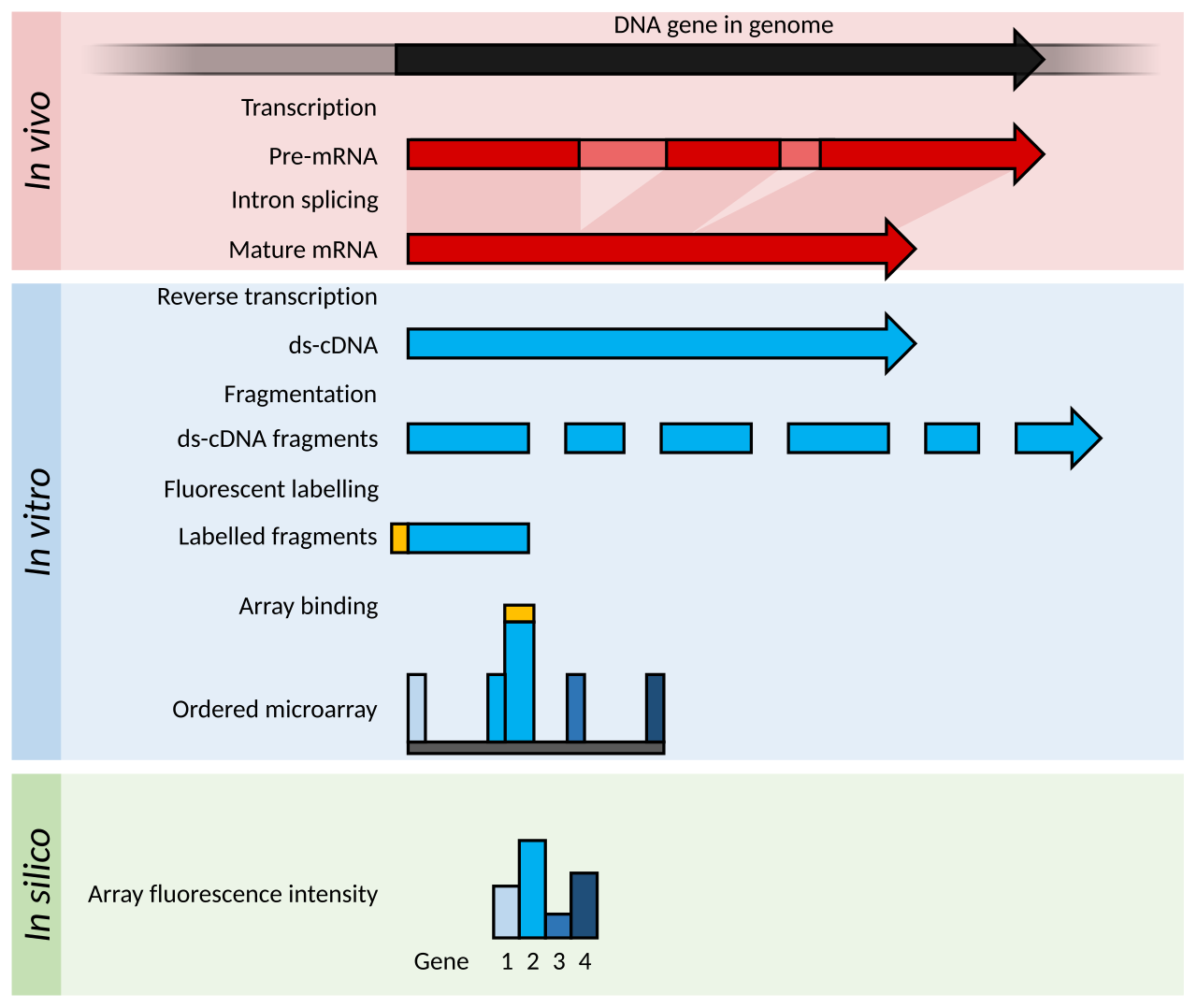
Principes et avancées
Les microarrays sont généralement constitués d'une grille d' oligomères nucléotidiques courts , appelés « sondes », généralement disposés sur une lame de verre. L'abondance des transcrits est déterminée par l'hybridation de transcrits marqués par fluorescence à ces sondes. L' intensité de fluorescence à chaque emplacement de sonde sur la matrice indique l'abondance des transcrits pour cette séquence de sonde. Les groupes de sondes conçus pour mesurer le même transcrit (c'est-à-dire l'hybridation d'un transcrit spécifique dans différentes positions) sont généralement appelés « ensembles de sondes ».
Les microarrays nécessitent certaines connaissances génomiques de l'organisme d'intérêt, par exemple sous la forme d'une séquence génomique annotée ou d'une bibliothèque d'EST qui peut être utilisée pour générer les sondes pour le réseau.
Méthodes
Les microarrays pour la transcriptomique appartiennent généralement à l'une des deux grandes catégories suivantes : les microarrays à faible densité ou les microarrays à sonde courte à haute densité. L'abondance des transcrits est déduite de l'intensité de la fluorescence dérivée des transcrits marqués par un fluorophore qui se lient au microarray.
Les matrices à faible densité tachetées comportent généralement des gouttes de l'ordre du picolitre d'une gamme d' ADNc purifiés disposés sur la surface d'une lame de verre. Ces sondes sont plus longues que celles des matrices à haute densité et ne peuvent pas identifier les événements d'épissage alternatifs . Les matrices à points utilisent deux fluorophores différents pour marquer les échantillons de test et de contrôle, et le rapport de fluorescence est utilisé pour calculer une mesure relative de l'abondance. Les matrices à haute densité utilisent un seul marqueur fluorescent, et chaque échantillon est hybridé et détecté individuellement. Les matrices à haute densité ont été popularisées par la matrice Affymetrix GeneChip , où chaque transcription est quantifiée par plusieurs sondes courtes de 25 -mer qui analysent ensemble un gène.
Les matrices NimbleGen étaient des matrices à haute densité produites par une méthode de photochimie sans masque , qui permettait une fabrication flexible de matrices en petit ou en grand nombre. Ces matrices comportaient des centaines de milliers de sondes de 45 à 85-mer et étaient hybridées avec un échantillon marqué d'une seule couleur pour l'analyse de l'expression. Certaines conceptions incorporaient jusqu'à 12 matrices indépendantes par lame.
ARN-Seq
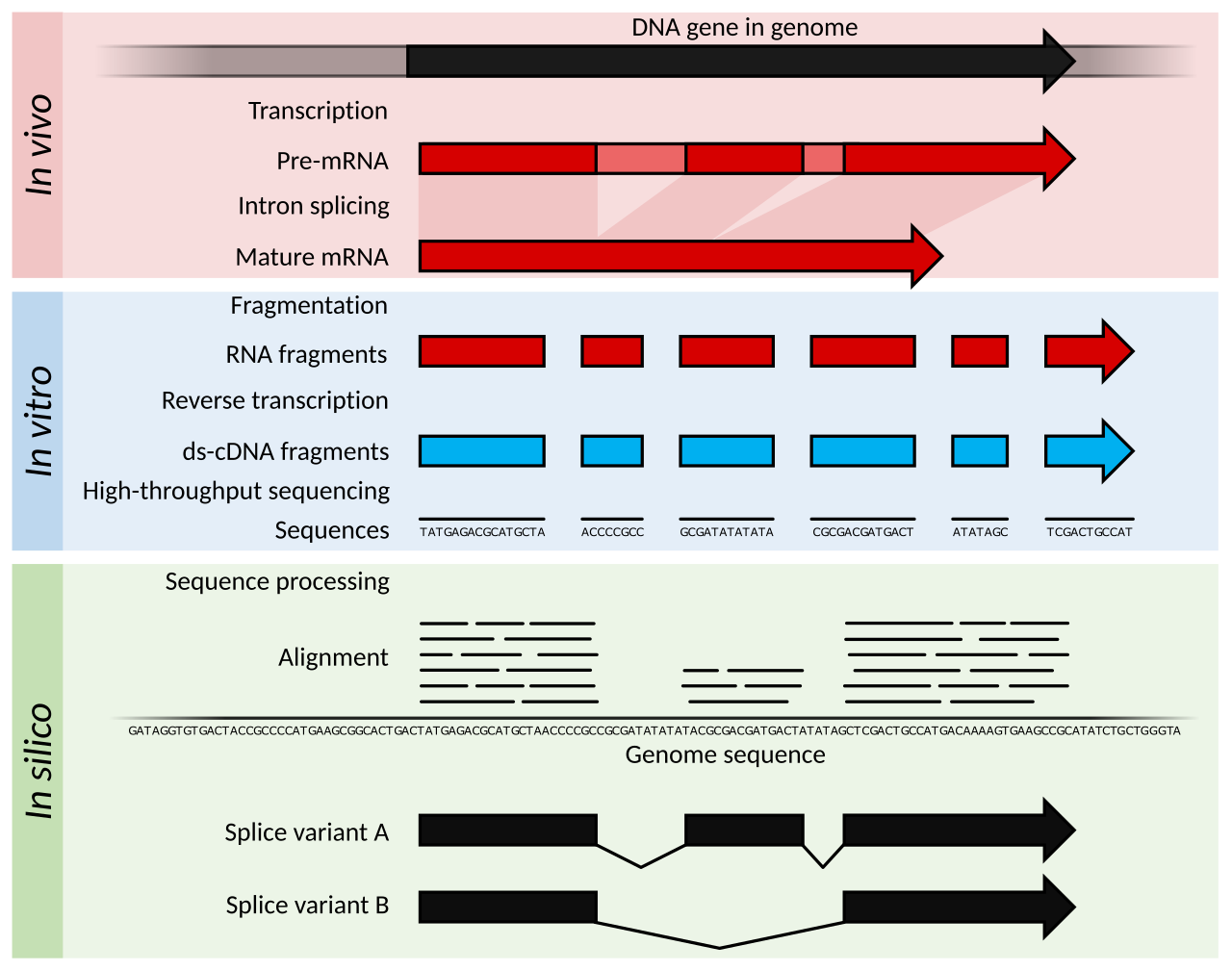
Principes et avancées
Français L'ARN-Seq fait référence à la combinaison d'une méthodologie de séquençage à haut débit avec des méthodes informatiques pour capturer et quantifier les transcrits présents dans un extrait d'ARN. Les séquences nucléotidiques générées ont généralement une longueur d'environ 100 pb, mais peuvent aller de 30 pb à plus de 10 000 pb selon la méthode de séquençage utilisée. L'ARN-Seq exploite un échantillonnage approfondi du transcriptome avec de nombreux fragments courts d'un transcriptome pour permettre la reconstruction informatique du transcrit d'ARN d'origine en alignant les lectures sur un génome de référence ou entre elles ( assemblage de novo ). Les ARN à faible et à forte abondance peuvent être quantifiés dans une expérience d'ARN-Seq ( plage dynamique de 5 ordres de grandeur ), un avantage clé par rapport aux transcriptomes de puces à ADN. De plus, les quantités d'ARN d'entrée sont beaucoup plus faibles pour le RNA-Seq (quantité nanogramme) par rapport aux microarrays (quantité microgramme), qui permettent l'examen du transcriptome même à une résolution unicellulaire lorsqu'il est combiné à l'amplification de l'ADNc. Théoriquement, il n'y a pas de limite supérieure de quantification dans le RNA-Seq, et le bruit de fond est très faible pour les lectures de 100 pb dans les régions non répétitives.
Le séquençage de l'ARN peut être utilisé pour identifier les gènes d'un génome ou pour identifier les gènes actifs à un moment donné. Le nombre de lectures peut également être utilisé pour modéliser avec précision le niveau d'expression relatif des gènes. La méthodologie du séquençage de l'ARN s'est constamment améliorée, principalement grâce au développement de technologies de séquençage de l'ADN pour augmenter le débit, la précision et la longueur de lecture. Depuis les premières descriptions en 2006 et 2008, le séquençage de l'ARN a été rapidement adopté et a dépassé les puces à ADN comme technique de transcriptomique dominante en 2015.
La quête de données sur le transcriptome au niveau des cellules individuelles a permis des avancées dans les méthodes de préparation des bibliothèques RNA-Seq, ce qui a entraîné des avancées spectaculaires en termes de sensibilité. Les transcriptomes monocellulaires sont désormais bien décrits et ont même été étendus au RNA-Seq in situ où les transcriptomes de cellules individuelles sont directement interrogés dans des tissus fixés .
Méthodes
Le séquençage de l'ARN a été mis au point en même temps que le développement rapide d'une gamme de technologies de séquençage de l'ADN à haut débit. Cependant, avant que les transcrits d'ARN extraits ne soient séquencés, plusieurs étapes de traitement clés sont réalisées. Les méthodes diffèrent dans l'utilisation de l'enrichissement des transcrits, de la fragmentation, de l'amplification, du séquençage simple ou apparié et de la préservation ou non des informations sur les brins.
La sensibilité d'une expérience de séquençage d'ARN peut être augmentée en enrichissant les classes d'ARN qui présentent un intérêt et en épuisant les ARN connus comme abondants. Les molécules d'ARNm peuvent être séparées à l'aide de sondes oligonucléotidiques qui lient leurs queues poly-A . Alternativement, la ribo-déplétion peut être utilisée pour éliminer spécifiquement les ARN ribosomiques (ARNr) abondants mais non informatifs par hybridation à des sondes adaptées aux séquences d'ARNr spécifiques du taxon (par exemple, l'ARNr de mammifère, l'ARNr de plante). Cependant, la ribo-déplétion peut également introduire un biais via l'épuisement non spécifique des transcrits hors cible. Les petits ARN, tels que les micro ARN , peuvent être purifiés en fonction de leur taille par électrophorèse sur gel et extraction.
Les ARNm étant plus longs que les longueurs de lecture des méthodes de séquençage à haut débit classiques, les transcriptions sont généralement fragmentées avant le séquençage. La méthode de fragmentation est un aspect clé de la construction d'une bibliothèque de séquençage. La fragmentation peut être réalisée par hydrolyse chimique , nébulisation , sonication ou transcription inverse avec des nucléotides de terminaison de chaîne . Alternativement, la fragmentation et le marquage de l'ADNc peuvent être effectués simultanément en utilisant des enzymes transposase .
Français Pendant la préparation du séquençage, les copies d'ADNc des transcrits peuvent être amplifiées par PCR pour enrichir les fragments qui contiennent les séquences d'adaptateur 5' et 3' attendues. L'amplification est également utilisée pour permettre le séquençage de très faibles quantités d'ARN en entrée, jusqu'à seulement 50 pg dans les applications extrêmes. Les contrôles de pointe d'ARN connus peuvent être utilisés pour l'évaluation du contrôle qualité afin de vérifier la préparation et le séquençage de la bibliothèque, en termes de teneur en GC , de longueur de fragment, ainsi que de biais dû à la position du fragment dans un transcrit. Les identificateurs moléculaires uniques (UMI) sont de courtes séquences aléatoires qui sont utilisées pour marquer individuellement les fragments de séquence pendant la préparation de la bibliothèque afin que chaque fragment marqué soit unique. Les UMI fournissent une échelle absolue pour la quantification, la possibilité de corriger le biais d'amplification ultérieur introduit pendant la construction de la bibliothèque et d'estimer avec précision la taille initiale de l'échantillon. Les UMI sont particulièrement bien adaptés à la transcriptomique de l'ARN-Seq à cellule unique, où la quantité d'ARN d'entrée est limitée et une amplification prolongée de l'échantillon est nécessaire.
Une fois les molécules de transcription préparées, elles peuvent être séquencées dans une seule direction (single-end) ou dans les deux directions (paired-end). Une séquence à une seule extrémité est généralement plus rapide à produire, moins chère que le séquençage à deux extrémités et suffisante pour quantifier les niveaux d'expression des gènes. Le séquençage à deux extrémités produit des alignements/assemblages plus robustes, ce qui est bénéfique pour l'annotation des gènes et la découverte des isoformes de transcription . Les méthodes d'ARN-Seq spécifiques à un brin préservent les informations sur le brin d'une transcription séquencée. Sans informations sur le brin, les lectures peuvent être alignées sur un locus de gène, mais n'indiquent pas dans quelle direction le gène est transcrit. Le séquençage d'ARN stranded est utile pour déchiffrer la transcription des gènes qui se chevauchent dans différentes directions et pour faire des prédictions génétiques plus robustes dans les organismes non modèles.
Légende : NCBI SRA – Archives de lecture de séquences d'informations du Centre national d'information sur la biotechnologie.
Actuellement, le séquençage de l'ARN repose sur la copie des molécules d'ARN dans les molécules d'ADNc avant le séquençage ; par conséquent, les plates-formes ultérieures sont les mêmes pour les données transcriptomiques et génomiques. Par conséquent, le développement des technologies de séquençage de l'ADN a été une caractéristique déterminante du séquençage de l'ARN. Le séquençage direct de l'ARN à l'aide du séquençage par nanopores représente une technique de pointe actuelle du séquençage de l'ARN. Le séquençage de l'ARN par nanopores peut détecter des bases modifiées qui seraient autrement masquées lors du séquençage de l'ADNc et élimine également les étapes d'amplification qui peuvent autrement introduire un biais.
La sensibilité et la précision d'une expérience de séquençage de l'ARN dépendent du nombre de lectures obtenues à partir de chaque échantillon. Un grand nombre de lectures est nécessaire pour assurer une couverture suffisante du transcriptome, permettant la détection de transcriptions à faible abondance. La conception expérimentale est encore compliquée par les technologies de séquençage avec une plage de sortie limitée, l'efficacité variable de la création de séquences et la qualité variable des séquences. À ces considérations s'ajoute le fait que chaque espèce possède un nombre différent de gènes et nécessite donc un rendement de séquence personnalisé pour un transcriptome efficace. Les premières études ont déterminé empiriquement des seuils appropriés, mais à mesure que la technologie a mûri, la couverture appropriée a été prédite par calcul par saturation du transcriptome. De manière quelque peu contre-intuitive, le moyen le plus efficace d'améliorer la détection de l'expression différentielle dans les gènes à faible expression est d'ajouter plus de réplicats biologiques plutôt que d'ajouter plus de lectures. Les critères de référence actuels recommandés par le projet Encyclopedia of DNA Elements (ENCODE) prévoient une couverture de l'exome de 70 fois pour l'ARN-Seq standard et une couverture de l'exome jusqu'à 500 fois pour détecter les transcrits et isoformes rares.
Analyse des données
Les méthodes de transcriptomique sont hautement parallèles et nécessitent des calculs importants pour produire des données significatives pour les expériences de microarray et d'ARN-Seq. Les données de microarray sont enregistrées sous forme d'images haute résolution , nécessitant une détection de caractéristiques et une analyse spectrale. Les fichiers d'images brutes de microarray ont chacun une taille d'environ 750 Mo, tandis que les intensités traitées ont une taille d'environ 60 Mo. Plusieurs sondes courtes correspondant à un seul transcrit peuvent révéler des détails sur la structure intron - exon , nécessitant des modèles statistiques pour déterminer l'authenticité du signal résultant. Les études d'ARN-Seq produisent des milliards de séquences d'ADN courtes, qui doivent être alignées sur des génomes de référence composés de millions à des milliards de paires de bases. L'assemblage de novo de lectures au sein d'un ensemble de données nécessite la construction de graphes de séquences très complexes . Les opérations RNA-Seq sont très répétitives et bénéficient d' un calcul parallélisé , mais les algorithmes modernes signifient que le matériel informatique grand public est suffisant pour des expériences de transcriptomique simples qui ne nécessitent pas d'assemblage de novo de lectures. Un transcriptome humain pourrait être capturé avec précision en utilisant RNA-Seq avec 30 millions de séquences de 100 pb par échantillon. Cet exemple nécessiterait environ 1,8 gigaoctet d'espace disque par échantillon lorsqu'il est stocké dans un format fastq compressé . Les données de comptage traitées pour chaque gène seraient beaucoup plus petites, équivalentes aux intensités de puces à ADN traitées. Les données de séquence peuvent être stockées dans des référentiels publics, tels que Sequence Read Archive (SRA). Les ensembles de données RNA-Seq peuvent être téléchargés via Gene Expression Omnibus.
Traitement d'image

Le traitement des images de microarray doit identifier correctement la grille régulière des caractéristiques d'une image et quantifier indépendamment l' intensité de fluorescence de chaque caractéristique. Les artéfacts d'image doivent également être identifiés et supprimés de l'analyse globale. Les intensités de fluorescence indiquent directement l'abondance de chaque séquence, puisque la séquence de chaque sonde sur le réseau est déjà connue.
Les premières étapes du séquençage de l'ARN comprennent également un traitement d'image similaire ; cependant, la conversion des images en données de séquence est généralement gérée automatiquement par le logiciel de l'instrument. La méthode de séquençage par synthèse d'Illumina produit un ensemble de clusters répartis sur la surface d'une cellule d'écoulement. La cellule d'écoulement est imagée jusqu'à quatre fois au cours de chaque cycle de séquençage, avec des dizaines à des centaines de cycles au total. Les clusters de cellules d'écoulement sont analogues aux spots de microarray et doivent être correctement identifiés au cours des premières étapes du processus de séquençage. Dans la méthode de pyroséquençage de Roche , l'intensité de la lumière émise détermine le nombre de nucléotides consécutifs dans une répétition d'homopolymère. Il existe de nombreuses variantes de ces méthodes, chacune avec un profil d'erreur différent pour les données résultantes.
Analyse des données RNA-Seq
Les expériences de séquençage de l'ARN génèrent un volume important de lectures de séquences brutes qui doivent être traitées pour produire des informations utiles. L'analyse des données nécessite généralement une combinaison d' outils logiciels bioinformatiques (voir également Liste des outils bioinformatiques de séquençage de l'ARN ) qui varient en fonction de la conception et des objectifs expérimentaux. Le processus peut être décomposé en quatre étapes : contrôle qualité, alignement, quantification et expression différentielle. Les programmes de séquençage de l'ARN les plus populaires sont exécutés à partir d'une interface de ligne de commande , soit dans un environnement Unix , soit dans l' environnement statistique R / Bioconductor .
Contrôle de qualité
Les lectures de séquences ne sont pas parfaites, il faut donc estimer la précision de chaque base de la séquence pour les analyses en aval. Les données brutes sont examinées pour garantir que : les scores de qualité des appels de base sont élevés, que le contenu en GC correspond à la distribution attendue, que les motifs de séquence courts ( k-mers ) ne sont pas surreprésentés et que le taux de duplication de lecture est suffisamment bas. Plusieurs options logicielles existent pour l'analyse de la qualité des séquences, notamment FastQC et FaQCs. Les anomalies peuvent être supprimées (élagage) ou marquées pour un traitement spécial lors des processus ultérieurs.
Alignement
Afin de lier l'abondance de lecture de séquence à l'expression d'un gène particulier, les séquences de transcription sont alignées sur un génome de référence ou alignées de novo les unes sur les autres si aucune référence n'est disponible. Les principaux défis pour les logiciels d'alignement comprennent une vitesse suffisante pour permettre l'alignement de milliards de séquences courtes dans un délai significatif, la flexibilité pour reconnaître et gérer l'épissage des introns de l'ARNm eucaryote et l'attribution correcte des lectures qui correspondent à plusieurs emplacements. Les progrès des logiciels ont largement abordé ces problèmes et l'augmentation de la longueur de lecture du séquençage réduit le risque d'alignements de lecture ambigus. Une liste des aligneurs de séquences à haut débit actuellement disponibles est tenue à jour par l' EBI .
L'alignement des séquences d'ARNm de transcription primaire dérivées d' eucaryotes sur un génome de référence nécessite une manipulation spécialisée des séquences d'introns , qui sont absentes de l'ARNm mature. Les aligneurs de lecture courts effectuent une série supplémentaire d'alignements spécifiquement conçus pour identifier les jonctions d'épissage , informés par les séquences canoniques des sites d'épissage et les informations connues sur les sites d'épissage des introns. L'identification des jonctions d'épissage des introns empêche les lectures d'être mal alignées sur les jonctions d'épissage ou rejetées par erreur, ce qui permet d'aligner davantage de lectures sur le génome de référence et d'améliorer la précision des estimations de l'expression des gènes. Étant donné que la régulation des gènes peut se produire au niveau de l' isoforme de l'ARNm , les alignements sensibles à l'épissage permettent également de détecter les changements d'abondance des isoformes qui seraient autrement perdus dans une analyse groupée.
L'assemblage de novo peut être utilisé pour aligner les lectures les unes sur les autres afin de construire des séquences de transcription complètes sans utiliser de génome de référence. Les défis particuliers à l'assemblage de novo comprennent des exigences de calcul plus importantes par rapport à un transcriptome basé sur une référence, une validation supplémentaire des variantes ou des fragments de gènes et une annotation supplémentaire des transcriptions assemblées. Les premières mesures utilisées pour décrire les assemblages de transcriptome, telles que N50 , se sont révélées trompeuses et des méthodes d'évaluation améliorées sont désormais disponibles. Les mesures basées sur l'annotation sont de meilleures évaluations de l'exhaustivité de l'assemblage, telles que le nombre de meilleurs résultats réciproques de contig . Une fois assemblé de novo , l'assemblage peut être utilisé comme référence pour les méthodes d'alignement de séquences ultérieures et l'analyse quantitative de l'expression génétique.
Légende : RAM – mémoire à accès aléatoire ; MPI – interface de passage de messages ; EST – balise de séquence exprimée.
Quantification
La quantification des alignements de séquences peut être effectuée au niveau du gène, de l'exon ou du transcrit. Les sorties typiques comprennent un tableau de nombres de lectures pour chaque caractéristique fournie au logiciel ; par exemple, pour les gènes dans un fichier de format de caractéristique générale . Les nombres de lectures de gènes et d'exons peuvent être calculés assez facilement en utilisant HTSeq, par exemple. La quantification au niveau du transcrit est plus compliquée et nécessite des méthodes probabilistes pour estimer l'abondance des isoformes du transcrit à partir d'informations de lecture courtes ; par exemple, en utilisant un logiciel de boutons de manchette. Les lectures qui s'alignent également bien sur plusieurs emplacements doivent être identifiées et soit supprimées, soit alignées sur l'un des emplacements possibles, soit alignées sur l'emplacement le plus probable.
Certaines méthodes de quantification peuvent contourner complètement la nécessité d'un alignement exact d'une lecture sur une séquence de référence. La méthode logicielle kallisto combine le pseudo-alignement et la quantification en une seule étape qui s'exécute 2 ordres de grandeur plus rapidement que les méthodes contemporaines telles que celles utilisées par le logiciel tophat/cufflinks, avec une charge de calcul moindre.
Expression différentielle
Une fois que les décomptes quantitatifs de chaque transcription sont disponibles, l'expression différentielle des gènes est mesurée en normalisant, en modélisant et en analysant statistiquement les données. La plupart des outils liront un tableau de gènes et liront les décomptes en entrée, mais certains programmes, tels que cuffdiff, accepteront les alignements de lecture au format de carte d'alignement binaire en entrée. Les résultats finaux de ces analyses sont des listes de gènes avec des tests par paires associés pour l'expression différentielle entre les traitements et les estimations de probabilité de ces différences.
Légende : ARNm - ARN messager.
Validation
Les analyses transcriptomiques peuvent être validées à l'aide d'une technique indépendante, par exemple, la PCR quantitative (qPCR), qui est reconnaissable et statistiquement évaluable. L'expression génétique est mesurée par rapport à des normes définies à la fois pour le gène d'intérêt et les gènes de contrôle . La mesure par qPCR est similaire à celle obtenue par RNA-Seq dans laquelle une valeur peut être calculée pour la concentration d'une région cible dans un échantillon donné. La qPCR est cependant limitée aux amplicons inférieurs à 300 pb, généralement vers l'extrémité 3' de la région codante, évitant le 3'UTR . Si la validation des isoformes de transcription est requise, une inspection des alignements de lecture RNA-Seq doit indiquer où les amorces qPCR peuvent être placées pour une discrimination maximale. La mesure de plusieurs gènes de contrôle avec les gènes d'intérêt produit une référence stable dans un contexte biologique. La validation qPCR des données RNA-Seq a généralement montré que différentes méthodes RNA-Seq sont fortement corrélées.
La validation fonctionnelle des gènes clés est une considération importante pour la planification post-transcriptomique. Les profils d'expression génétique observés peuvent être fonctionnellement liés à un phénotype par une étude indépendante de knock-down / sauvetage dans l'organisme d'intérêt.
Applications
Diagnostic et profilage des maladies
Les stratégies transcriptomiques ont été largement appliquées dans divers domaines de la recherche biomédicale, notamment le diagnostic et le profilage des maladies . Les approches RNA-Seq ont permis l'identification à grande échelle des sites de démarrage de la transcription , la découverte d'une utilisation alternative des promoteurs et de nouvelles altérations de l'épissage . Ces éléments régulateurs sont importants dans les maladies humaines et, par conséquent, la définition de ces variantes est cruciale pour l'interprétation des études d'association aux maladies . L'ARN-Seq peut également identifier les polymorphismes mononucléotidiques (SNP) associés aux maladies, l'expression spécifique des allèles et les fusions de gènes , ce qui contribue à la compréhension des variantes causales des maladies.
Les rétrotransposons sont des éléments transposables qui prolifèrent dans les génomes eucaryotes par un processus impliquant la transcription inverse . L'ARN-Seq peut fournir des informations sur la transcription des rétrotransposons endogènes qui peuvent influencer la transcription des gènes voisins par divers mécanismes épigénétiques qui conduisent à la maladie. De même, le potentiel d'utilisation de l'ARN-Seq pour comprendre les maladies liées au système immunitaire se développe rapidement en raison de la capacité à disséquer les populations de cellules immunitaires et à séquencer les répertoires de récepteurs des cellules T et B des patients.
Transcriptomes humains et pathogènes
L'ARN-Seq des agents pathogènes humains est devenu une méthode établie pour quantifier les changements d'expression génétique, identifier de nouveaux facteurs de virulence , prédire la résistance aux antibiotiques et dévoiler les interactions immunitaires hôte-pathogène . L'un des principaux objectifs de cette technologie est de développer des mesures optimisées de contrôle des infections et un traitement individualisé ciblé .
L'analyse transcriptomique s'est principalement concentrée sur l'hôte ou sur le pathogène. Le double séquençage de l'ARN a été appliqué pour profiler simultanément l'expression de l'ARN dans le pathogène et l'hôte tout au long du processus d'infection. Cette technique permet d'étudier la réponse dynamique et les réseaux de régulation des gènes inter-espèces chez les deux partenaires d'interaction, du contact initial jusqu'à l'invasion et la persistance finale du pathogène ou son élimination par le système immunitaire de l'hôte.
Réponses à l'environnement
La transcriptomique permet d'identifier les gènes et les voies qui répondent aux stress environnementaux biotiques et abiotiques et les neutralisent . La nature non ciblée de la transcriptomique permet l'identification de nouveaux réseaux transcriptionnels dans des systèmes complexes. Par exemple, l'analyse comparative d'une gamme de lignées de pois chiches à différents stades de développement a identifié des profils transcriptionnels distincts associés aux stress de sécheresse et de salinité , y compris l'identification du rôle des isoformes de transcription de AP2 - EREBP . L' étude de l'expression des gènes pendant la formation du biofilm par le pathogène fongique Candida albicans a révélé un ensemble de gènes co-régulés essentiels à l'établissement et au maintien du biofilm.
Le profilage transcriptomique fournit également des informations cruciales sur les mécanismes de résistance aux médicaments . L'analyse de plus de 1000 isolats de Plasmodium falciparum , un parasite virulent responsable du paludisme chez l'homme, a permis d'identifier que la régulation positive de la réponse protéique dépliée et la progression plus lente au cours des premiers stades du cycle de développement intraérythrocytaire asexué étaient associées à la résistance à l'artémisinine dans les isolats d' Asie du Sud-Est .
L'utilisation de la transcriptomique est également importante pour étudier les réponses dans l'environnement marin. En écologie marine, le « stress » et l'« adaptation » ont été parmi les sujets de recherche les plus courants, en particulier en ce qui concerne le stress anthropique, comme le changement global et la pollution . La plupart des études dans ce domaine ont été réalisées sur des animaux , bien que les invertébrés aient été sous-représentés. Un problème persiste : le manque d'études génétiques fonctionnelles, qui entravent les annotations génétiques , en particulier pour les espèces non modèles, et peuvent conduire à des conclusions vagues sur les effets des réponses étudiées.
Annotation de la fonction des gènes
Toutes les techniques transcriptomiques se sont révélées particulièrement utiles pour identifier les fonctions des gènes et identifier les responsables de phénotypes particuliers. La transcriptomique des écotypes d'Arabidopsis qui hyperaccumulent les métaux a mis en corrélation les gènes impliqués dans l'absorption des métaux , la tolérance et l'homéostasie avec le phénotype. L'intégration d'ensembles de données RNA-Seq dans différents tissus a été utilisée pour améliorer l'annotation des fonctions des gènes dans des organismes d'importance commerciale (par exemple le concombre ) ou des espèces menacées (par exemple le koala ).
L'assemblage des lectures RNA-Seq ne dépend pas d'un génome de référence et est donc idéal pour les études d'expression génétique d'organismes non modèles avec des ressources génomiques inexistantes ou peu développées. Par exemple, une base de données de SNP utilisée dans les programmes de sélection de sapins de Douglas a été créée par analyse de novo du transcriptome en l'absence d'un génome séquencé . De même, les gènes qui fonctionnent dans le développement des tissus cardiaques, musculaires et nerveux chez les homards ont été identifiés en comparant les transcriptomes des différents types de tissus sans utiliser de séquence génomique. des régions codantes de protéines jusqu'alors inconnues dans des génomes séquencés existants.
ARN non codant
La transcriptomique est généralement appliquée au contenu en ARNm de la cellule. Cependant, les mêmes techniques sont également applicables aux ARN non codants (ARNnc) qui ne sont pas traduits en protéine, mais qui ont des fonctions directes (par exemple, des rôles dans la traduction des protéines , la réplication de l'ADN , l'épissage de l'ARN et la régulation transcriptionnelle ). Beaucoup de ces ARNnc affectent les états pathologiques, notamment le cancer, les maladies cardiovasculaires et neurologiques.
Bases de données du transcriptome
Les études de transcriptomique génèrent de grandes quantités de données dont les applications potentielles vont bien au-delà des objectifs initiaux d'une expérience. Ainsi, les données brutes ou traitées peuvent être déposées dans des bases de données publiques pour garantir leur utilité pour la communauté scientifique au sens large. Par exemple, en 2018, le Gene Expression Omnibus contenait des millions d'expériences.
Légende : NCBI – Centre national d'information sur la biotechnologie ; EBI – Institut européen de bioinformatique ; DDBJ – Banque de données ADN du Japon ; ENA – Archives européennes des nucléotides ; MIAME – Informations minimales sur une expérience de microarray ; MINSEQE – Informations minimales sur une expérience de séquençage de nucléotides à haut débit.