La validation de la structure macromoléculaire est le processus d'évaluation de la fiabilité des modèles atomiques tridimensionnels de grandes molécules biologiques telles que les protéines et les acides nucléiques . Ces modèles, qui fournissent les coordonnées 3D de chaque atome de la molécule (voir exemple sur l'image), proviennent d' expériences de biologie structurale comme la cristallographie aux rayons X ou la résonance magnétique nucléaire (RMN) . La validation comporte trois aspects : 1) vérifier la validité des milliers, voire des millions, de mesures réalisées lors de l'expérience ; 2) vérifier la cohérence du modèle atomique avec ces données expérimentales ; et 3) vérifier la cohérence du modèle avec les propriétés physico-chimiques connues.
Les protéines et les acides nucléiques sont les piliers de la biologie, assurant les réactions chimiques nécessaires, l'organisation structurale, la croissance, la mobilité, la reproduction et la sensibilité à l'environnement. Leurs fonctions biologiques reposent sur la structure tridimensionnelle détaillée des molécules et la maîtrise de leurs modifications. Pour comprendre et contrôler ces fonctions, il est indispensable de connaître précisément les modèles qui représentent ces structures, leurs nombreux atouts comme leurs rares faiblesses.
Les utilisateurs finaux des modèles macromoléculaires comprennent les cliniciens, les enseignants et les étudiants, ainsi que les biologistes structuraux eux-mêmes, les rédacteurs et les relecteurs de revues scientifiques , les expérimentateurs étudiant les macromolécules par d'autres techniques, et les théoriciens et bioinformaticiens étudiant les propriétés plus générales des molécules biologiques. Leurs intérêts et leurs besoins varient, mais tous bénéficient grandement d'une compréhension globale et locale de la fiabilité des modèles.
La cristallographie macromoléculaire a été précédée par le domaine plus ancien de la cristallographie aux rayons X des petites molécules (pour les structures comportant moins de quelques centaines d'atomes). Les données de diffraction des petites molécules atteignent une résolution bien supérieure à celle des macromolécules et présentent une relation mathématique très précise avec le modèle atomique. Le résidu, ou facteur R, mesure la concordance entre les données expérimentales et les valeurs rétrocalculées à partir du modèle atomique. Pour une structure de petite molécule bien déterminée, le facteur R est presque aussi faible que l'incertitude sur les données expérimentales (nettement inférieure à 5 %). Par conséquent, ce test unique fournit à lui seul la majeure partie de la validation nécessaire, mais un certain nombre de contrôles supplémentaires de cohérence et de méthodologie sont effectués par un logiciel automatisé comme exigence pour les articles de structure cristalline de petites molécules soumis aux revues de l' Union internationale de cristallographie (IUCr) telles que Acta Crystallographica section B ou C. Les coordonnées atomiques de ces structures de petites molécules sont archivées et accessibles via la Cambridge Structural Database (CSD) ou la Crystallography Open Database (COD).Le premier logiciel de validation macromoléculaire a été développé vers 1990 pour les protéines. Il comprenait une validation croisée Rfree pour l'adéquation du modèle aux données , les paramètres de longueur et d'angle de liaison pour la géométrie covalente , et des critères conformationnels des chaînes latérales et du squelette peptidique . Pour les structures macromoléculaires, les modèles atomiques sont déposés dans la Protein Data Bank (PDB), qui demeure l'unique archive de ces données. La PDB a été créée dans les années 1970 au Brookhaven National Laboratory , puis en 2000 au RCSB (Research Collaboration for Structural Biology) de l' université Rutgers [ , et étendue en 2003 pour devenir la wwPDB ( Worldwide Protein Data Bank ) avec l'ajout de sites d'accès en Europe.) et l'Asie (), et avec des données RMN traitées à la BioMagResBank (BMRB) dans le Wisconsin.
Validation rapidly became standard in the field, with further developments described below. *Obviously needs expansion*
A large boost was given to the applicability of comprehensive validation for both x-ray and NMR as of February 1, 2008, when the worldwide Protein Data Bank (wwPDB) made mandatory the deposition of experimental data along with atomic coordinates. Since 2012 strong forms of validation have been in the process of being adopted for wwPDB deposition from recommendations of the wwPDB Validation Task Force committees for x-ray crystallography, for NMR, for SAXS (small-angle x-ray scattering), and for cryoEM (cryo-Electron Microscopy).
Stages of validation
Validations can be broken into three stages: validating the raw data collected (data validation), the interpretation of the data into the atomic model (model-to-data validation), and finally validation on the model itself. While the first two steps are specific to the technique used, validating the arrangement of atoms in the final model is not.
Model validation
Geometry
Conformation (dihedrals): protein & RNA
The backbone and side-chain dihedral angles of protein and RNA have been shown to have specific combinations of angles which are allowed (or forbidden). For protein backbone dihedrals (φ, ψ), this has been addressed by the legendary Ramachandran Plot while for side-chain dihedrals (χ's), one should refer to the Dunbrack Backbone-dependent rotamer library.
Bien que les ARNm soient généralement éphémères et monocaténaires, il existe une abondance d'ARN non codants présentant différents repliements secondaires et tertiaires (ARNt, ARNr, etc.) et contenant une prépondérance de paires de bases canoniques Watson-Crick (WC), ainsi qu'un nombre significatif de paires de bases non-Watson-Crick (NWC). Ces ARN sont donc également soumis à une validation structurale classique, comme pour les hélices d'acides nucléiques. La pratique courante consiste à analyser les paramètres géométriques intra- (transnationaux : décalage, glissement, élévation ; rotationnels : inclinaison, roulis, torsion) et inter-paires de bases (transnationaux : cisaillement, décalage, étirement ; rotationnels : flambage, hélice, ouverture) afin de déterminer s'ils se situent dans les intervalles de valeurs suggérées. Ces paramètres décrivent les orientations relatives des deux paires de bases appariées l'une par rapport à l'autre dans les deux brins (intra) ainsi que celles des deux paires de bases empilées (inter) l'une par rapport à l'autre. Ensemble, ils permettent donc de valider les structures d'acides nucléiques en général. Étant donné que les hélices d'ARN sont de petite taille (en moyenne : 10 à 20 pb), l'utilisation du potentiel électrostatique de surface comme paramètre de validation s'est avérée avantageuse, notamment pour la modélisation.
Empilement et électrostatique : protéines globulaires
Pour les protéines globulaires, l'organisation atomique interne (résultant d'interactions locales à courte portée) des chaînes latérales s'avère cruciale pour la stabilisation structurale du repliement protéique. Par ailleurs, l'harmonie électrostatique (interactions non locales à longue portée) du repliement global est également essentielle à sa stabilisation. Les anomalies d'organisation comprennent les encombrements stériques , les lacunes et les cavités , tandis que la disharmonie électrostatique fait référence à des charges partielles déséquilibrées au sein du cœur protéique (particulièrement pertinentes pour les intérieurs de protéines conçus). Si le score d'encombrement de Molprobity identifie les encombrements stériques avec une très haute résolution, le diagramme de complémentarité combine les anomalies d'organisation avec le déséquilibre électrostatique des chaînes latérales et les signaux relatifs à l'un ou l'autre de ces phénomènes, voire aux deux.
Glucides
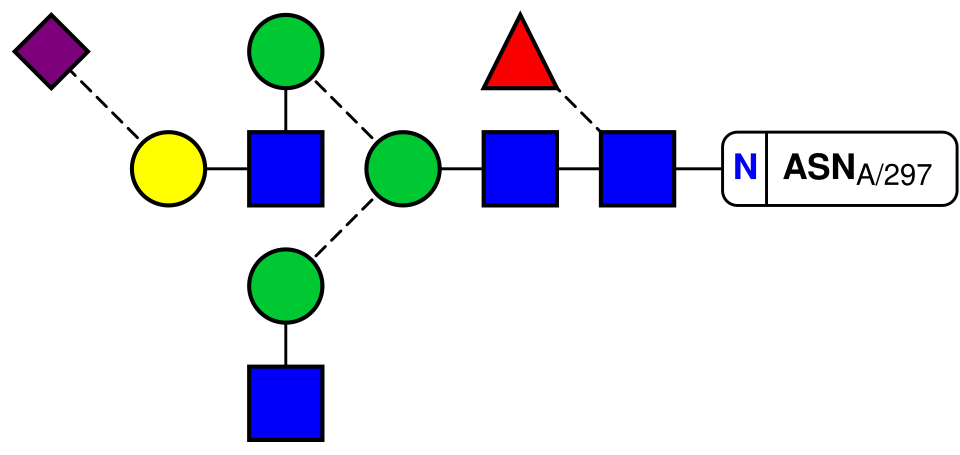
Privateer génère également des diagrammes SVG bidimensionnels évolutifs, conformes à la nomenclature symbolique standard de l'ouvrage Essentials of Glycobiology et contenant toutes les informations de validation sous forme d'infobulles (voir figure). Cette fonctionnalité est actuellement intégrée à d'autres programmes CCP4, tels que le programme de visualisation moléculaire CCP4mg (via la représentation 3D Glycoblocks , conforme à la nomenclature symbolique standard ) et l'interface graphique de la suite, CCP4i2.
Validation pour la cristallographie

Relation avec la résolution et le facteur B
Validation des données
Facteurs de structure
Jumelage
Validation du modèle par rapport aux données
Résidus et Rfree
Corrélation dans l'espace réel
Amélioration par la correction des problèmes diagnostiqués
En résonance magnétique nucléaire
Validation des données : déplacements chimiques, NOE, RDC
- AVS
- La suite de validation d'attribution ( AVS ) vérifie la liste des déplacements chimiques au format BioMagResBank (BMRB) pour détecter les problèmes.
- PSVS
- Serveur de validation de la structure des protéines au NESG basé sur des statistiques de récupération d'informations
- PROCESSUS
- PROSESS (Protein Structure Evaluation Suite & Server) est un nouveau serveur web qui propose une évaluation des modèles structuraux de protéines par déplacements chimiques RMN ainsi que par NOE, paramètres géométriques et basés sur les connaissances.
- LACS
- L'analyse linéaire des déplacements chimiques est utilisée pour le référencement absolu des données de déplacement chimique.
Validation du modèle par rapport aux données
TALOS+ prédit les angles de torsion du squelette protéique à partir de données de déplacement chimique. Fréquemment utilisé pour générer des contraintes supplémentaires appliquées à un modèle de structure lors de son affinement.
Validation du modèle : comme ci-dessus

Dynamique : domaines centraux vs boucles, queues et domaines mobiles
L'un des besoins essentiels pour la validation d'ensembles structuraux par RMN est de distinguer les régions bien caractérisées (celles pour lesquelles on dispose de données expérimentales) des régions très mobiles et/ou pour lesquelles aucune donnée n'a été observée. Plusieurs méthodes, actuelles ou proposées, permettent d'effectuer cette distinction, comme l'indice de pelote statistique , mais la communauté RMN n'a pas encore adopté de méthode standardisée.
Logiciels et sites web
En cryo-ME
La cryo-microscopie électronique présente des défis particuliers pour les concepteurs de modèles, car la densité électronique observée est souvent insuffisante pour résoudre les atomes individuels, ce qui entraîne une probabilité d'erreurs plus élevée.
Des outils de validation géométrique similaires à ceux utilisés en cristallographie aux rayons X peuvent servir à mettre en évidence les choix de modélisation invraisemblables et à orienter le modélisateur vers des structures plus proches de l'état natif. La méthode CaBLAM, qui utilise uniquement les atomes Cα convient aux structures à basse résolution obtenues par cryo-microscopie électronique
Une méthode de calcul de la carte de densité différentielle a été formulée pour la cryo-microscopie électronique. Une validation croisée utilisant une carte « libre », comparable à l’utilisation d’un facteur R libre , est également disponible. Parmi les autres méthodes de vérification de l’adéquation du modèle à la carte, on peut citer les coefficients de corrélation, le FSC du modèle à la carte, les cartes de confiance, CryoEF (vérification du biais d’orientation) et TEMPy SMOC.
En SAXS
La diffusion des rayons X aux petits angles (SAXS) est un domaine de la détermination structurale en pleine expansion, à la fois comme source d'approximation de la structure 3D pour les cas initiaux ou complexes et comme composante des méthodes hybrides de détermination structurale, en combinaison avec la RMN, la microscopie électronique, la cristallographie, la réticulation ou les calculs informatiques. Le développement de normes de validation fiables pour l'interprétation des données SAXS et la qualité des modèles obtenus suscite un vif intérêt, mais aucune méthode n'est encore largement utilisée. Trois avancées récentes dans ce sens sont la création d'un groupe de travail sur la validation de la diffusion aux petits angles par la Protein Data Bank mondiale et son rapport initial , un ensemble de normes suggérées pour l'inclusion des données dans les publications et une première proposition de critères statistiques pour l'évaluation automatisée de la qualité
Pour la biologie computationnelle
Il est difficile de valider de manière pertinente un modèle macromoléculaire individuel, purement computationnel, en l'absence de données expérimentales pour cette molécule, car le modèle présentant la meilleure géométrie et le meilleur score conformationnel n'est pas nécessairement celui qui se rapproche le plus de la réponse exacte. Par conséquent, la validation de la modélisation computationnelle repose en grande partie sur l'évaluation des méthodes. Afin d'éviter les biais et les raisonnements fallacieux, des concours de prédiction en double aveugle ont été mis en place. Le premier exemple en est le CASP (Critical Assessment of Structure Prediction), organisé tous les deux ans depuis 1994, qui évalue les prédictions de structures protéiques 3D à partir de structures cristallographiques ou RMN nouvellement résolues et dont la confidentialité est maintenue jusqu'à la fin du concours. Le critère principal d'évaluation du CASP est un score pondéré, appelé GDT-TS, qui évalue la concordance des positions des atomes Cα entre les modèles prédits et expérimentaux.